8. Creating the Gene Table
A copy of this notebook is available on the PyModulon GitHub page: Creating the Gene Table Notebook
First, download the FASTA and GFF files for your organism and its plasmids from NCBI. We will use the E. coli K-12 MG1655 strain as an example, which does not have plasmids.
[2]:
from pymodulon import example_data
8.1. Get information from GFF files
[3]:
from pymodulon.gene_util import *
Enter the location of all your GFF files here:
[4]:
gff_files = [example_data.ecoli_gff]
The following cell will convert all the GFF files into a single Pandas DataFrame for easy manipulation. Pseudogenes have multiple rows in a GFF file (one for each fragment), but only the first fragment will be kept.
[5]:
keep_cols = ['accession','start','end','strand','gene_name','old_locus_tag','gene_product','ncbi_protein']
DF_annot = gff2pandas(gff_files,index='locus_tag')
DF_annot = DF_annot[keep_cols]
DF_annot.head()
WARNING:root:Duplicate locus_tag detected. Dropping duplicates.
[5]:
| accession | start | end | strand | gene_name | old_locus_tag | gene_product | ncbi_protein | |
|---|---|---|---|---|---|---|---|---|
| locus_tag | ||||||||
| b0001 | NC_000913.3 | 190 | 255 | + | thrL | None | thr operon leader peptide | NP_414542.1 |
| b0002 | NC_000913.3 | 337 | 2799 | + | thrA | None | fused aspartate kinase/homoserine dehydrogenase 1 | NP_414543.1 |
| b0003 | NC_000913.3 | 2801 | 3733 | + | thrB | None | homoserine kinase | NP_414544.1 |
| b0004 | NC_000913.3 | 3734 | 5020 | + | thrC | None | threonine synthase | NP_414545.1 |
| b0005 | NC_000913.3 | 5234 | 5530 | + | yaaX | None | DUF2502 domain-containing protein YaaX | NP_414546.1 |
To ensure that the gene index used is identical to the expression matrix, load in your data.
[6]:
DF_log_tpm = example_data.load_example_log_tpm()
DF_log_tpm.head()
[6]:
| ecoli_00001 | ecoli_00002 | ecoli_00009 | ecoli_00010 | |
|---|---|---|---|---|
| Geneid | ||||
| b0001 | 10.473721 | 10.271944 | 10.315476 | 10.808135 |
| b0002 | 10.260569 | 10.368555 | 10.735874 | 10.726916 |
| b0003 | 9.920277 | 10.044224 | 10.528432 | 10.503092 |
| b0004 | 9.936694 | 10.010638 | 9.739519 | 9.722997 |
| b0005 | 7.027515 | 7.237449 | 6.745798 | 6.497823 |
Check that the genes are the same in the expression dataset as in the annotation dataframe. Mismatched genes are listed below.
[7]:
test = DF_annot.sort_index().index == DF_log_tpm.sort_index().index
DF_annot[~test]
[7]:
| accession | start | end | strand | gene_name | old_locus_tag | gene_product | ncbi_protein | |
|---|---|---|---|---|---|---|---|---|
| locus_tag |
8.2. (Optional) KEGG and COGs
8.2.1. Generate nucleotide fasta files for CDS
Enter the location of all your fasta files here:
[8]:
fasta_files = [example_data.ecoli_fasta]
The following code generates CDS files using your FASTA and GFF3 files
[9]:
from Bio import SeqIO
cds_list = []
for fasta in fasta_files:
seq = SeqIO.read(fasta,'fasta')
# Get gene information for genes in this fasta file
df_genes = DF_annot[DF_annot.accession == seq.id]
for i,row in df_genes.iterrows():
cds = seq[row.start-1:row.end]
if row.strand == '-':
cds = seq[row.start-1:row.end].reverse_complement()
cds.id = row.name
cds.description = row.gene_name if pd.notnull(row.gene_name) else row.name
cds_list.append(cds)
[10]:
cds_list[:5]
[10]:
[SeqRecord(seq=Seq('ATGAAACGCATTAGCACCACCATTACCACCACCATCACCATTACCACAGGTAAC...TGA', SingleLetterAlphabet()), id='b0001', name='NC_000913.3', description='thrL', dbxrefs=[]),
SeqRecord(seq=Seq('ATGCGAGTGTTGAAGTTCGGCGGTACATCAGTGGCAAATGCAGAACGTTTTCTG...TGA', SingleLetterAlphabet()), id='b0002', name='NC_000913.3', description='thrA', dbxrefs=[]),
SeqRecord(seq=Seq('ATGGTTAAAGTTTATGCCCCGGCTTCCAGTGCCAATATGAGCGTCGGGTTTGAT...TAA', SingleLetterAlphabet()), id='b0003', name='NC_000913.3', description='thrB', dbxrefs=[]),
SeqRecord(seq=Seq('ATGAAACTCTACAATCTGAAAGATCACAACGAGCAGGTCAGCTTTGCGCAAGCC...TAA', SingleLetterAlphabet()), id='b0004', name='NC_000913.3', description='thrC', dbxrefs=[]),
SeqRecord(seq=Seq('GTGAAAAAGATGCAATCTATCGTACTCGCACTTTCCCTGGTTCTGGTCGCTCCC...TAA', SingleLetterAlphabet()), id='b0005', name='NC_000913.3', description='yaaX', dbxrefs=[])]
Uncomment the line below to write the CDS file
[11]:
# SeqIO.write(cds_list,'CDS.fna','fasta')
8.2.2. Run EggNOG Mapper
Upload the CDS.fna file from your organism directory (within the sequence_files folder)
Make sure to limit the taxonomy to the correct level
After the job is submitted, you must follow the link in your email to run the job.
Once the job completes (after ~4 hrs), download the annotations file.
Save the annotation file
8.2.3. Get KEGG IDs
Once you have the EggNOG annotations, load the annotation file
[12]:
eggnog_file = example_data.ecoli_eggnog
[13]:
DF_eggnog = pd.read_csv(eggnog_file,sep='\t',skiprows=4,header=None)
eggnog_cols = ['query_name','seed eggNOG ortholog','seed ortholog evalue','seed ortholog score',
'Predicted taxonomic group','Predicted protein name','Gene Ontology terms',
'EC number','KEGG_orth','KEGG_pathway','KEGG_module','KEGG_reaction',
'KEGG_rclass','BRITE','KEGG_TC','CAZy','BiGG Reaction','tax_scope',
'eggNOG OGs','bestOG_deprecated','COG','eggNOG free text description']
DF_eggnog.columns = eggnog_cols
# Strip last three rows as they are comments
DF_eggnog = DF_eggnog.iloc[:-3]
# Set locus tag as index
DF_eggnog = DF_eggnog.set_index('query_name')
DF_eggnog.index.name = 'locus_tag'
DF_eggnog.head()
[13]:
| seed eggNOG ortholog | seed ortholog evalue | seed ortholog score | Predicted taxonomic group | Predicted protein name | Gene Ontology terms | EC number | KEGG_orth | KEGG_pathway | KEGG_module | ... | KEGG_rclass | BRITE | KEGG_TC | CAZy | BiGG Reaction | tax_scope | eggNOG OGs | bestOG_deprecated | COG | eggNOG free text description | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| locus_tag | |||||||||||||||||||||
| b0002 | 316407.85674276 | 0.000000e+00 | 1600.5 | Escherichia | thrA | GO:0003674,GO:0003824,GO:0004072,GO:0004412,GO... | 1.1.1.3,2.7.2.4 | ko:K12524 | ko00260,ko00261,ko00270,ko00300,ko01100,ko0111... | M00016,M00017,M00018,M00526,M00527 | ... | RC00002,RC00043,RC00087 | ko00000,ko00001,ko00002,ko01000 | NaN | NaN | iECDH1ME8569_1439.ECDH1ME8569_0002,iEcDH1_1363... | Escherichia | 1MW3H@1224,1RN1G@1236,3XN2A@561,COG0460@1,COG0... | NA|NA|NA | E | homoserine dehydrogenase I |
| b0003 | 316407.85674277 | 3.000000e-178 | 630.9 | Escherichia | thrB | GO:0000096,GO:0000097,GO:0003674,GO:0003824,GO... | 2.7.1.39 | ko:K00872 | ko00260,ko01100,ko01110,ko01120,ko01230,map002... | M00018 | ... | RC00002,RC00017 | ko00000,ko00001,ko00002,ko01000 | NaN | NaN | iECSE_1348.ECSE_0003 | Escherichia | 1MW8I@1224,1RMYR@1236,3XN01@561,COG0083@1,COG0... | NA|NA|NA | F | Catalyzes the ATP-dependent phosphorylation of... |
| b0004 | 316407.21321894 | 1.200000e-246 | 858.6 | Escherichia | thrC | GO:0003674,GO:0003824,GO:0004795,GO:0005575,GO... | 4.2.3.1 | ko:K01733 | ko00260,ko00750,ko01100,ko01110,ko01120,ko0123... | M00018 | ... | RC00017,RC00526 | ko00000,ko00001,ko00002,ko01000 | NaN | NaN | iLF82_1304.LF82_2261,iNRG857_1313.NRG857_00025 | Escherichia | 1MUWQ@1224,1RQ0H@1236,3XP31@561,COG0498@1,COG0... | NA|NA|NA | E | Catalyzes the gamma-elimination of phosphate f... |
| b0005 | 316407.21321895 | 1.400000e-23 | 115.5 | Escherichia | yaaX | NaN | NaN | NaN | NaN | NaN | ... | NaN | NaN | NaN | NaN | NaN | Escherichia | 1N4SV@1224,1S9KR@1236,2B58A@1,32XJS@2,3XRGB@561 | NA|NA|NA | S | Protein of unknown function (DUF2502) |
| b0006 | 198214.SF0006 | 1.600000e-143 | 515.4 | Gammaproteobacteria | NaN | NaN | NaN | NaN | NaN | NaN | ... | NaN | NaN | NaN | NaN | NaN | Escherichia | 1MUAF@1224,1RMTD@1236,COG3022@1,COG3022@2 | NA|NA|NA | S | Belongs to the UPF0246 family |
5 rows × 21 columns
Now we will pull the KEGG information from the eggNOG file, including orthology, pathway, module, and reactions for each gene.
[14]:
DF_kegg = DF_eggnog[['KEGG_orth','KEGG_pathway','KEGG_module','KEGG_reaction']]
# Melt dataframe
DF_kegg = DF_kegg.reset_index().melt(id_vars='locus_tag')
# Remove null values
DF_kegg = DF_kegg[DF_kegg.value.notnull()]
# Split comma-separated values into their own rows
list2struct = []
for name,row in DF_kegg.iterrows():
for val in row.value.split(','):
list2struct.append([row.locus_tag,row.variable,val])
DF_kegg = pd.DataFrame(list2struct,columns=['gene_id','database','kegg_id'])
# Remove ko entries, as only map entries are searchable in KEGG pathway
DF_kegg = DF_kegg[~DF_kegg.kegg_id.str.startswith('ko')]
DF_kegg.head()
[14]:
| gene_id | database | kegg_id | |
|---|---|---|---|
| 2750 | b0002 | KEGG_pathway | map00260 |
| 2751 | b0002 | KEGG_pathway | map00261 |
| 2752 | b0002 | KEGG_pathway | map00270 |
| 2753 | b0002 | KEGG_pathway | map00300 |
| 2754 | b0002 | KEGG_pathway | map01100 |
8.2.4. Save KEGG information
Uncomment the line below to save the KEGG file
[15]:
# DF_kegg.to_csv('kegg_mapping.csv')
8.2.5. Save COGs to annotation dataframe
[16]:
DF_annot['COG'] = DF_eggnog.COG
# Make sure COG only has one entry per gene
DF_annot['COG'] = [item[0] if isinstance(item,str) else item for item in DF_annot['COG']]
8.3. Uniprot ID mapping
The uniprot_id_mapping function is a python wrapper for the Uniprot ID mapping tool. Use input_id=P_REFSEQ_AC if the FASTA/GFFf files are from RefSeq, and input_id=EMBL if the files are from Genbank.
[17]:
mapping_uniprot = uniprot_id_mapping(DF_annot.ncbi_protein.fillna(''),input_id='P_REFSEQ_AC',output_id='ACC',
input_name='ncbi_protein',output_name='uniprot')
mapping_uniprot.head()
[17]:
| ncbi_protein | uniprot | |
|---|---|---|
| 2090 | NP_416515.1 | A0A0G3HGF9 |
| 4381 | NP_418733.1 | A0A0N9YI43 |
| 3651 | NP_418014.1 | A0A223DQZ5 |
| 26 | NP_414563.1 | A0A385XJ53 |
| 1978 | NP_416408.1 | A0A385XJ53 |
[18]:
# Merge with current annotation
DF_annot = pd.merge(DF_annot.reset_index(),mapping_uniprot,how='left',on='ncbi_protein')
DF_annot.set_index('locus_tag',inplace=True)
DF_annot.head()
[18]:
| accession | start | end | strand | gene_name | old_locus_tag | gene_product | ncbi_protein | COG | uniprot | |
|---|---|---|---|---|---|---|---|---|---|---|
| locus_tag | ||||||||||
| b0001 | NC_000913.3 | 190 | 255 | + | thrL | None | thr operon leader peptide | NP_414542.1 | NaN | P0AD86 |
| b0002 | NC_000913.3 | 337 | 2799 | + | thrA | None | fused aspartate kinase/homoserine dehydrogenase 1 | NP_414543.1 | E | P00561 |
| b0003 | NC_000913.3 | 2801 | 3733 | + | thrB | None | homoserine kinase | NP_414544.1 | F | P00547 |
| b0004 | NC_000913.3 | 3734 | 5020 | + | thrC | None | threonine synthase | NP_414545.1 | E | P00934 |
| b0005 | NC_000913.3 | 5234 | 5530 | + | yaaX | None | DUF2502 domain-containing protein YaaX | NP_414546.1 | S | P75616 |
8.4. Add Biocyc Operon information
To obtain operon information from Biocyc, follow the steps below
Go to Biocyc.org (you may need to create an account and/or login)
Change the organism database to your organism/strain
Select SmartTables -> Special SmartTables
Select “All genes of <organism>”
Select the “Gene Name” column
Under “ADD TRANSFORM COLUMN” select “Genes in same transcription unit”
Select the “Genes in same transcription unit” column
Under “ADD PROPERTY COLUMN” select “Accession-1”
Under OPERATIONS, select “Export” -> “to Spreadsheet File…”
Select “common names” and click “Export smarttable”
Add file location below and run the code cell
[19]:
biocyc_file = example_data.ecoli_biocyc
DF_biocyc = pd.read_csv(biocyc_file,sep='\t')
# Remove genes with no accession
DF_biocyc = DF_biocyc[DF_biocyc['Accession-1'].notnull()]
# Set the accession (i.e. locus tag) as index
DF_biocyc = DF_biocyc.set_index('Accession-1').sort_values('Left-End-Position')
# Only keep genes in the final annotation file
DF_biocyc = DF_biocyc.reindex(DF_annot.index)
# Reformat transcription units
DF_biocyc['operon_list'] = DF_biocyc['Accession-1.1'].apply(reformat_biocyc_tu)
DF_biocyc.head()
[19]:
| Gene Name | Left-End-Position | Right-End-Position | Product | Genes in same transcription unit | Accession-1.1 | operon_list | |
|---|---|---|---|---|---|---|---|
| locus_tag | |||||||
| b0001 | thrL | 190.0 | 255.0 | <i>thr</i> operon leader peptide | thrL // thrA // thrB // thrC | b0001 // b0002 // b0003 // b0004 | b0001;b0002;b0003;b0004 |
| b0002 | thrA | 337.0 | 2799.0 | fused aspartate kinase/homoserine dehydrogenase 1 | thrA // thrB // thrC // thrL | b0002 // b0003 // b0004 // b0001 | b0001;b0002;b0003;b0004 |
| b0003 | thrB | 2801.0 | 3733.0 | homoserine kinase | thrA // thrB // thrC // thrL | b0002 // b0003 // b0004 // b0001 | b0001;b0002;b0003;b0004 |
| b0004 | thrC | 3734.0 | 5020.0 | threonine synthase | thrA // thrB // thrC // thrL | b0002 // b0003 // b0004 // b0001 | b0001;b0002;b0003;b0004 |
| b0005 | yaaX | 5234.0 | 5530.0 | DUF2502 domain-containing protein YaaX | yaaX | b0005 | b0005 |
8.4.1. Assign unique IDs to operons
The following code assigns unique names to each operon
[20]:
# Get all operons
operons = DF_biocyc['operon_list'].unique()
# Map each operon to a unique string
operon_dict = {operon: "Op"+str(i) for i, operon in enumerate(operons)}
# Add names to dataframe
DF_biocyc['operon'] = [operon_dict[op] for op in DF_biocyc["operon_list"]]
DF_biocyc.head()
[20]:
| Gene Name | Left-End-Position | Right-End-Position | Product | Genes in same transcription unit | Accession-1.1 | operon_list | operon | |
|---|---|---|---|---|---|---|---|---|
| locus_tag | ||||||||
| b0001 | thrL | 190.0 | 255.0 | <i>thr</i> operon leader peptide | thrL // thrA // thrB // thrC | b0001 // b0002 // b0003 // b0004 | b0001;b0002;b0003;b0004 | Op0 |
| b0002 | thrA | 337.0 | 2799.0 | fused aspartate kinase/homoserine dehydrogenase 1 | thrA // thrB // thrC // thrL | b0002 // b0003 // b0004 // b0001 | b0001;b0002;b0003;b0004 | Op0 |
| b0003 | thrB | 2801.0 | 3733.0 | homoserine kinase | thrA // thrB // thrC // thrL | b0002 // b0003 // b0004 // b0001 | b0001;b0002;b0003;b0004 | Op0 |
| b0004 | thrC | 3734.0 | 5020.0 | threonine synthase | thrA // thrB // thrC // thrL | b0002 // b0003 // b0004 // b0001 | b0001;b0002;b0003;b0004 | Op0 |
| b0005 | yaaX | 5234.0 | 5530.0 | DUF2502 domain-containing protein YaaX | yaaX | b0005 | b0005 | Op1 |
Finally, merge the Biocyc information with the main annotation DataFrame
[21]:
DF_annot['operon'] = DF_biocyc['operon']
8.5. Clean up and save annotation
First, we will re-order the annotation columns
[22]:
if 'old_locus_tag' in DF_annot.columns:
order = ['gene_name','accession','old_locus_tag','start','end','strand','gene_product','COG','uniprot','operon']
else:
order = ['gene_name','accession','start','end','strand','gene_product','COG','uniprot','operon']
DF_annot = DF_annot[order]
[23]:
DF_annot.head()
[23]:
| gene_name | accession | old_locus_tag | start | end | strand | gene_product | COG | uniprot | operon | |
|---|---|---|---|---|---|---|---|---|---|---|
| locus_tag | ||||||||||
| b0001 | thrL | NC_000913.3 | None | 190 | 255 | + | thr operon leader peptide | NaN | P0AD86 | Op0 |
| b0002 | thrA | NC_000913.3 | None | 337 | 2799 | + | fused aspartate kinase/homoserine dehydrogenase 1 | E | P00561 | Op0 |
| b0003 | thrB | NC_000913.3 | None | 2801 | 3733 | + | homoserine kinase | F | P00547 | Op0 |
| b0004 | thrC | NC_000913.3 | None | 3734 | 5020 | + | threonine synthase | E | P00934 | Op0 |
| b0005 | yaaX | NC_000913.3 | None | 5234 | 5530 | + | DUF2502 domain-containing protein YaaX | S | P75616 | Op1 |
8.6. Final statistics
The following graphs show how much information is available for the organism.
[24]:
import seaborn as sns
import matplotlib.pyplot as plt
sns.set_style('ticks')
[25]:
fig,ax = plt.subplots()
DF_annot.count().plot(kind='bar',ax=ax)
ax.set_ylabel('# of Values',fontsize=18)
ax.tick_params(labelsize=16)
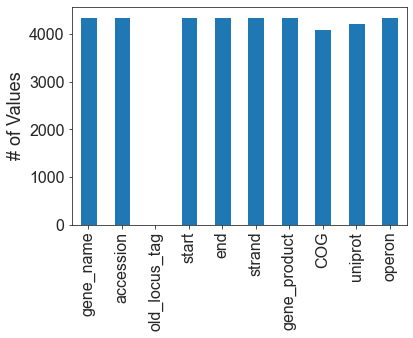
8.7. Fill missing values
Some organisms are missing gene names, so these will be filled with locus tag gene names.
[26]:
# Fill in missing gene names with locus tag names
DF_annot['tmp_name'] = DF_annot.copy().index.tolist()
DF_annot.gene_name.fillna(DF_annot.tmp_name,inplace=True)
DF_annot.drop('tmp_name',axis=1,inplace=True)
COG letters will also be converted to the full name.
[27]:
# Fill missing COGs with X
DF_annot['COG'].fillna('X',inplace=True)
# Change single letter COG annotation to full description
DF_annot['COG'] = DF_annot.COG.apply(cog2str)
counts = DF_annot.COG.value_counts()
plt.pie(counts.values,labels=counts.index);
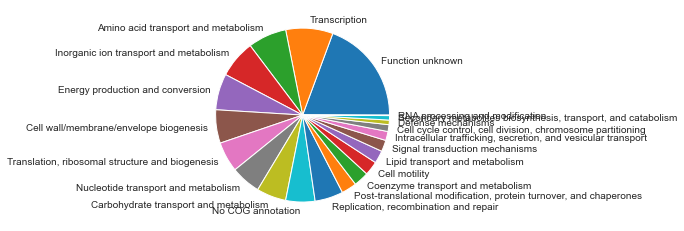
Uncomment the following line to save the gene annotation dataset
[28]:
# DF_annot.to_csv('gene_info.csv')
8.8. GO Annotations
To start, download the GO Annotations for your organism from AmiGO 2
Go to AmiGO 2
Filter for your organism
Click
CustomDLDrag
GO class (direct)to the end of your Selected FieldsEnter the location of your GO annotation file below and run the following code block
[29]:
go_file = example_data.ecoli_go_example
[30]:
DF_GO = pd.read_csv(go_file,sep='\t',header=None,usecols=[2,10,17])
DF_GO.columns = ['gene_name','gene_id','gene_ontology']
DF_GO.gene_id.fillna(DF_GO.gene_name,inplace=True)
DF_GO = DF_GO[['gene_id','gene_ontology']]
DF_GO.head()
[30]:
| gene_id | gene_ontology | |
|---|---|---|
| 0 | b0312|ECK0310 | betaine-aldehyde dehydrogenase activity |
| 1 | b0312|ECK0310 | betaine-aldehyde dehydrogenase activity |
| 2 | b0312|ECK0310 | betaine-aldehyde dehydrogenase activity |
| 3 | b0312|ECK0310 | betaine-aldehyde dehydrogenase activity |
| 4 | b0312|ECK0310 | response to osmotic stress |
Take a look at the gene_id column: 1. Make sure there are no null entries 2. Check which naming convention is used (locus tag, new locus tag, or gene name)
If it looks like it uses the old locus tag, set old_locus_tag to True.
In this example, the gene_id column needs to be altered slightly to match with the gene name. This may not be necessary for your organism.
[31]:
def format_go_gene_id(gene_id):
locus_tags = [item for item in gene_id.split('|') if re.match('b\d{4}',item)]
if len(locus_tags) > 0:
return locus_tags[0]
else:
return None
DF_GO.gene_id = DF_GO.gene_id.apply(format_go_gene_id)
Now we remove null entries
[32]:
DF_GO = DF_GO[DF_GO.gene_id.notnull()]
[33]:
naming = 'locus_tag' # Can be "gene_name" or "old_locus_tag"
if naming != 'locus_tag':
convert_tags = {value:key for key,value in DF_annot[naming].items()}
DF_GO.gene_id = DF_GO.gene_id.apply(lambda x: convert_tags[x])
[34]:
DF_GO.head()
[34]:
| gene_id | gene_ontology | |
|---|---|---|
| 0 | b0312 | betaine-aldehyde dehydrogenase activity |
| 1 | b0312 | betaine-aldehyde dehydrogenase activity |
| 2 | b0312 | betaine-aldehyde dehydrogenase activity |
| 3 | b0312 | betaine-aldehyde dehydrogenase activity |
| 4 | b0312 | response to osmotic stress |
Uncomment the line below to save the annotations
[35]:
# DF_GO[['gene_id','gene_ontology']].to_csv('GO_annotations.csv')